Inspired by Sudoku, U of T researchers create novel protein-folding algorithm for drug discovery
Published: October 1, 2020
Computational biologists at the University of Toronto’s Donnelly Centre for Cellular and Biomolecular Research have developed an artificial intelligence algorithm that has the potential to create novel protein molecules as finely tuned therapeutics.
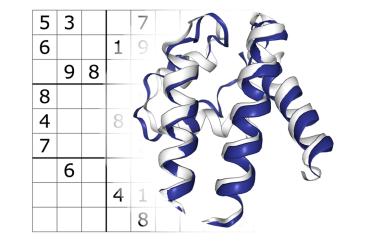
The team led by Philip M. Kim, a professor of molecular genetics in U of T’s Temerty Faculty of Medicine and of computer science in the Faculty of Arts & Science, has developed ProteinSolver, a graph neural network that can design a fully new protein to fit a given geometric shape. The researchers took inspiration from the Japanese number puzzle Sudoku, whose constraints are conceptually similar to those of a protein molecule.
Their findings are published in the journal Cell Systems.
“The parallel with Sudoku becomes apparent when you depict a protein molecule as a network,” says Kim, adding that the portrayal of proteins in graph form is standard practice in computational biology.
A newly synthesized protein is a string of amino-acids, stitched together according to the instructions in that protein’s gene code. The amino-acid polymer then folds in and around itself into a three-dimensional molecular machine that can be harnessed for medicine.
A protein converted into a graph looks like a network of nodes, representing amino-acids that are connected by edges, which are the distances between them within the molecule. By applying principles from graph theory, it then becomes possible to model the molecule’s geometry for a specific purpose to, for example, neutralize an invading virus or shut down an overactive receptor in cancer.
Proteins make good drugs thanks to the three-dimensional features on their surface with which they bind to cellular targets with more precision than the synthetic small molecule drugs that tend to be broad spectrum and can lead to harmful side effects.
Just over a third of all medications approved over the last few years are proteins, which also make up the vast majority of top ten drugs globally, Kim says. Insulin, antibodies and growth factors are just a few examples of injectable cellular proteins, also known as biologics, that are already in use.
However, designing proteins from scratch remains incredibly difficult, owing to the vast number of possible structures to choose from.
“The main problem in protein design is that you have a very large search space,” says Kim, referring to the many ways in which the 20 naturally occurring amino-acids can be combined into protein structures.
“For a standard-length protein of 100 amino-acids, there are 20 to the power of 100 possible molecular structures – that’s more than the number of molecules in the universe,” he says.
Kim decided to turn the problem on its head by starting with a three-dimensional structure and working out its amino acid composition.
“It’s the protein design, or the inverse protein folding problem: You have a shape in mind and you want a sequence (of amino-acids) that will fold into that shape. Solving this is in some ways more useful than protein folding, as you can in theory generate new proteins for any purpose,” says Kim.
That’s when Alexey Strokach, a PhD student in Kim’s lab, turned to Sudoku after learning about its relatedness to molecular geometry in a class.
In Sudoku, the goal is to find missing values in a sparsely filled grid by observing a set of rules and the existing number values.
Individual amino-acids in a protein molecule are similarly constrained by their neighbours. Local electrostatic forces ensure that amino-acids carrying opposite electric charge pack closely together while those with the same charge are pulled apart.
Strokach first built the constraints found in Sudoku into a neural network algorithm. He then trained the algorithms on a vast database of available protein structures and their amino-acid sequences. The goal was to teach the algorithm, ProteinSolver, the rules – honed by evolution over millions of years – that govern packing amino acids together into smaller folds. Applying these rules to the engineering process should increase the chances of having a functional protein at the end.
The researchers then tested ProteinSolver by giving it existing protein folds and asking it to generate amino acid sequences that can build them. They then took the novel computed sequences, which do not exist in nature, and manufactured the corresponding protein variants in the lab. The variants folded into the expected structures, showing that the approach works.
In its current form, ProteinSolver is able to compute novel amino acid sequences for any protein fold known to be geometrically stable. But the ultimate goal is to engineer novel protein structures with entirely new biological functions, as new therapeutics, for example.
“The ultimate goal is for someone to be able to draw a completely new protein by hand and compute sequences for that, and that’s what we are working on now,” says Strokach.
The researchers made ProteinSolver and the code behind it open source and available to the wider research community through a user-friendly website.
The research received support from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada.


